Fold tree
KEYWORDS: CORE_CONCEPTS GENERAL
Tutorial by Parisa Hosseinzadeh (parisah@uw.edu). Edited by Sebastian Rämisch (raemisch@scripps.edu). File created 21 June 2016 as part of the 2016 Documentation eXtreme Rosetta Workshop (XRW).
In this tutorial you will learn about the concept of fold tree, internal coordinates, and how to use them wisely to obtain meaningful outputs.
Internal Coordinates
If you open a PDB file and look at it, you can see that it has the information of all the atoms with their (x,y,z) coordinates in 3D space. When you want to move a protein, you can change all 3 coordinates of each atom. In other words, each atom has 3 degrees of freedom. However, 3 degrees of freedom is simply too much for our calculations. In order to make things simpler, Rosetta uses internal coordinates instead. You can imagine that another way of defining an atom uniquely is to use the bond, angle, and torsion of it with regards to its neighbor atoms. When you use bond, angle, and torsion angle values of residues, instead of the x,y,z values, to describe an atom, you are using its internal coordinates.
In the internal coordinate world, you move objects by changing the bond, angle, and torsions, so there are again 3 degrees of freedom. However, these degrees of freedom are not equally important. For most protein modeling, there will be very little change in the bond lengths and angles - effectively, we can consider them to be fixed. The motions are mostly happening by a change in the torsion angles. Hence, we are reducing the degrees of freedom from 3 per atom to 1 per atom. Additionally, for certain atoms (like most hydrogens) their torsional degree of freedom is fixed by their chemical environment. For these atoms the degrees of freedom have been reduced from 3 to 0.
If you go to
<path-to-Rosetta>/main/database/chemical/residue_type_sets/fa_standard/residue_types/l-caayou can find several params files for all 20 amino acids. Open one and look at it. The lines started with ICOOR_INTERNAL show the internal coordinates of the atoms in that residue (see here for details of what each column in ICOOR_INTERNAL line means.)
The Lever Arm Effect
When you change the torsion angles in a given sets of residues but you set the rest to not move, what will happen? Let's see the simple scheme below:
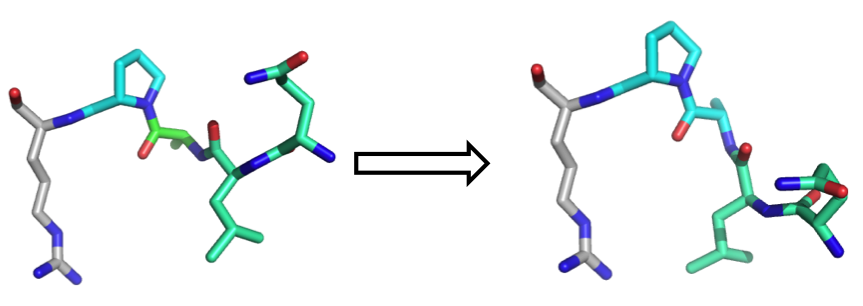
You can see that in order to change one specific torsion angle in the cyan residue, all of the atoms that are to the right of that change should all move. In other words, although they are held fixed "WITH RESPECT TO EACH OTHER", they still move together to accommodate changes in the downstream cyan residue.
Now let's look at the same example, but with a small difference:
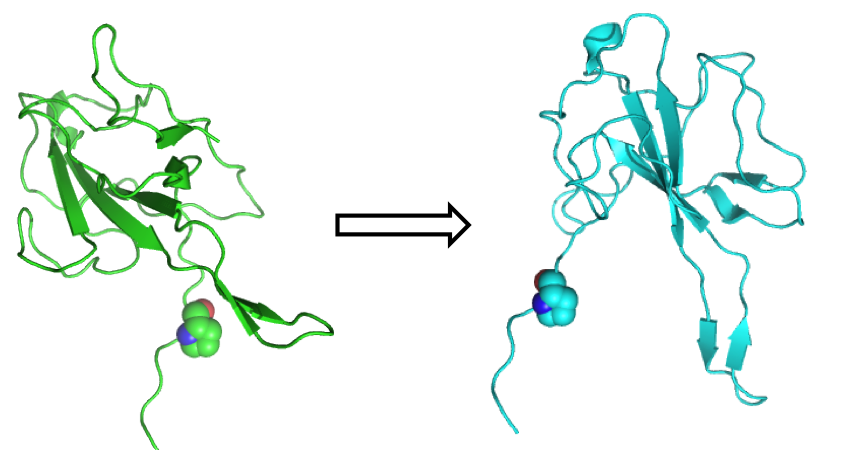
You can see that a small change in one torsion angle at the N-terminus of the protein can cause huge movements in the rest of the protein. This is called the lever arm effect.
How can we avoid this?
Using the Fold Tree
In this part we see how to use the fold tree to control movements of the parts in a protein with respects to each other. The fold tree is a way to tell Rosetta the connectivity between residues in a given structure. It defines what residues are UPSTREAM or parents and what residues are DOWNSTREAM or children. Let's go through some examples to make things clear.
In this example, you will be using the structure of the N-terminal part of nucleocapsid from coronaviruse. Navigate to the fold-tree directory in tutorials using this command:
> cd <path_to_Rosetta_directory>/demos/tutorials/fold_treeOpen the capsid.pdb file provided for you in the inputs directory. You can see that it has a long, unstructured N-terminal and the rest of the structure is mostly beta sheets. We know that the N-terminal region is disordered, so before working with the PDB, we want to relax just that part.
The cps_relax1.xml is a Rosetta script provided to you that relaxes residues 1-20 of the structure using a MoveMap.
...
<MoveMap name="part">
<Span begin=1 end=20 bb=1 chi=1/>
</MoveMap>
...It also uses the default Rosetta fold tree, which is shown in caps_tree1.ft file in the input directory:
FOLD_TREE EDGE 1 133 -1-> This reads as: "Edge from 1 to 133, which is a polymeric edge (-1)"
You start from residue 1 in chain A and go all the way to the last residue (133 in this example) of the chain. So any movements in the upstream residues (residues that are close to N-terminus) will cause movements in residues downstream (closer to C-terminal). This chain is a non-disrupted continuous cluster of residues that are connected by covalent bonds in a linear way. We call this cluster an EDGE. -1 means that the residues in the EDGE are connected through covalent bonds.
In your fold_tree folder, run the script using the command below to relax residues 1-20 without changing the backbone in the remainder of the structure:
$> <path_to_Rosetta_directory>/main/source/bin/rosetta_scripts.default.linuxgccrelease -in:file:s inputs/capsid.pdb -parser:protocol inputs/caps_relax1.xml -out:prefix test1_NOTE: you may need to change your executable from what is provided here depending on your compilation.
After 1-10 minutes, you can see that the output (test1_capsid_0001.pdb) is generated.
Now compare the original structure with the output (e.g. align them in Pymol). You can see that the N-terminal part has moved drastically compared to the original structure. If we align only the N-terminal parts, you can see that the rest of the protein has moved drastically to accommodate changes in the small 20 residue N-terminal.
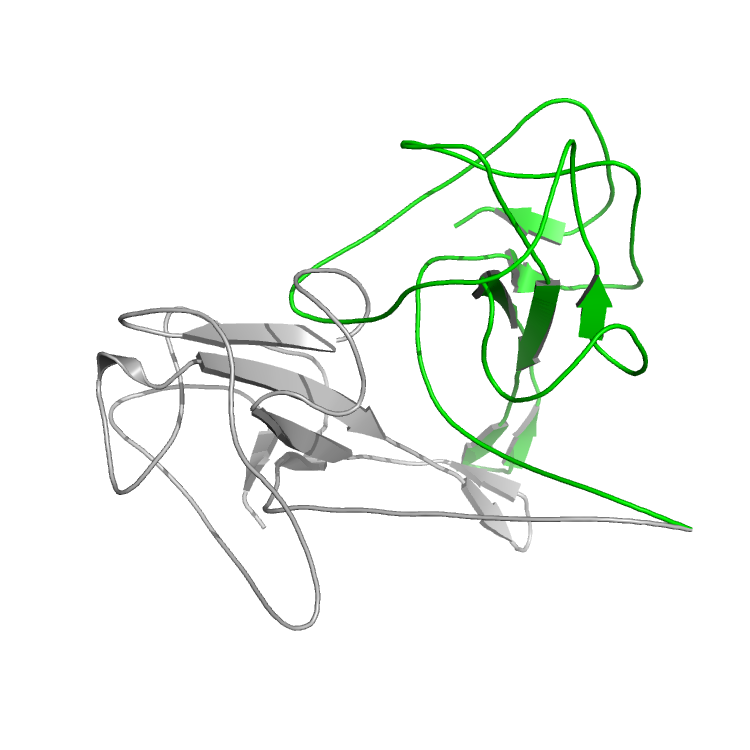
The original structure is in grey and the test1 structure is shown in green.
Now, let's see what happens when we swap the position of the downstream and upstream, telling Rosetta that movements should propagate from C to N terminal. This is shown in caps_tree2.ft file:
FOLD_TREE EDGE 133 1 -1You can see that the fold tree is directional.
let's run one round of relax on the capsid.pdb structure but this time with the new fold tree:
$> ../../../main/source/bin/rosetta_scripts.default.linuxgccrelease -in:file:s inputs/capsid.pdb -parser:protocol inputs/caps_relax2.xml -out:prefix test2_This shouldn't take longer than 10 min. Look at the test2_capsid_0001.pdb output structure and compare it with both the capsid.pdb and the test1_capsid_0001.pdb structure. (the two outputs are provided in the outputs directory). You can see now that the movements in the protein are much smaller. This is because now the C-terminal parts are upstream of where the changes are happening and are not moving as a result of movements in the N-terminal.

The original structure is in grey and the test2 structure is shown in cyan.
So if you have a protein with very flexible N-terminal that will move a lot during your run, you may want to change the fold tree.
Complexes With More Than One Chain and Jumps
Now let's run another example. In this example we are running a hypothetical ubiquitin dimer. The pdb is called ubq_dimer.pdb in the inputs directory.
Let's see what happens if I relax the pose. In the ubq_relax1.xml Rosetta script we used a movemap to fix the backbone except for the small loop in the C-terminal. In the first run, we will use the default fold tree of Rosetta for protein complexes. Let's take a look at ubq_tree1.ft that contains this fold tree:
FOLD_TREE EDGE 1 76 -1 EDGE 76 77 1 EDGE 77 152 -1-> This reads as: "Edge from 1 to 76, which is a protein edge (-1); Edge form 76 to 77, which is the first jump edge (1)"; Edge from 77 to 152, which is again a protein edge (-1)"
You can see that we have three EDGEs. The first one should be familiar. It says that the first edge starts from residue 1 and goes through residue 76 by covalent linkage (basically, chain A). The third one is doing the same thing but for the chain B. (Note that these are pose numberings.)
Now, let's look at the second edge. You can see that it goes from the last residue of chain A (76) to the first residue of chain B (77). This is not a covalent interaction! But, because Rosetta uses internal coordinates (torsion angles), and not just 3D-coordinates, all pieces in a structure have to be connected. If there is no covalent connection (like in our case here), chains have to be connected with an imaginary link that we call a JUMP. A jump is an edge by itself. In a fold tree, jumps are shown as positive numbers. The first jump gets the number 1 and the numbers go up for each jump.
Now let's relax our dimer using the default fold tree. Run this command in your fold-tree directory:
$> ../../../main/source/bin/rosetta_scripts.default.linuxgccrelease -s inputs/ubq_dimer.pdb -parser:protocol inputs/ubq_relax1.xml -out:prefix test1_After 1-5 min, you should have an output called test1_ubq_dimer_0001.pdb. Take a look at the structure and compare it to the original dimer. You can see that the second chain moved a lot from it's original position. This is another example of the lever arm effect. The movements in the last few residues in the C_terminal of chain A has propagated all the way to chain A.
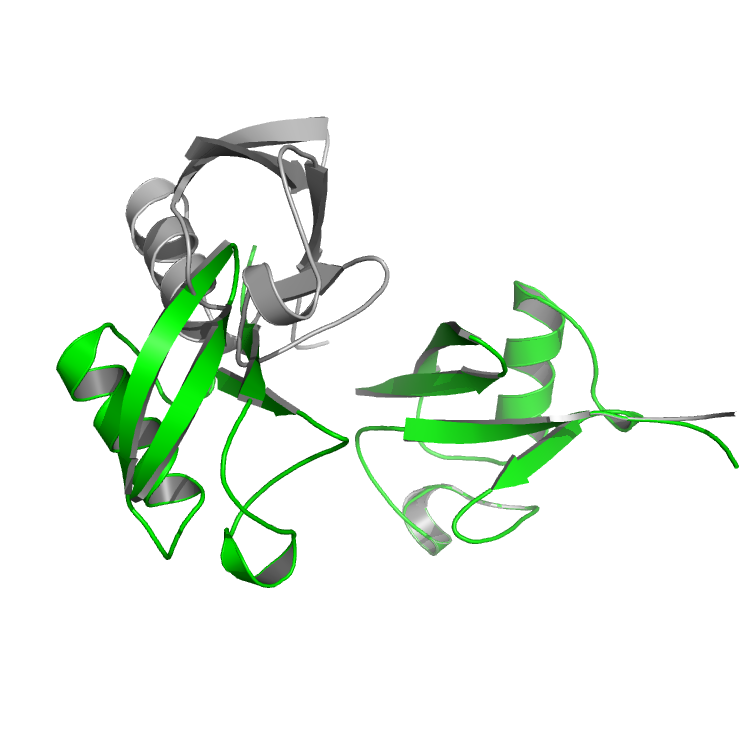
The original structure is in grey and the test1 structure is shown in green.
Now, let's see how we can fix it. Say we know based on experiments that the Val71 in chain A and Val146 in chain B are important in the interface formation and want to make sure they stay close during relaxation. So, we can re-define our fold tree so that residues 71 and 146 are the immediate parents, or the most upstream. Take a look at the modified fold tree (inputs/ubq_tree2.ft)
FOLD_TREE EDGE 71 1 -1 EDGE 71 76 -1 EDGE 71 146 1 EDGE 146 77 -1 EDGE 146 152 -1-> as before, this is a protein egde (-1), protein edge (-1), jump edge (1), protein edge (-1), protein edge (-1)
You can see that now there are multiple EDGEs. Chain A is now defined by two edges: the first one goes through from residue 71 all the way back to N-terminal of chain A and the second one goes from residue 71 to C-terminal of chain A. The JUMP is now from 71 to 146, the two parents. And chain B is also defined by two edges. The scheme below shows how the two fold tree differ:
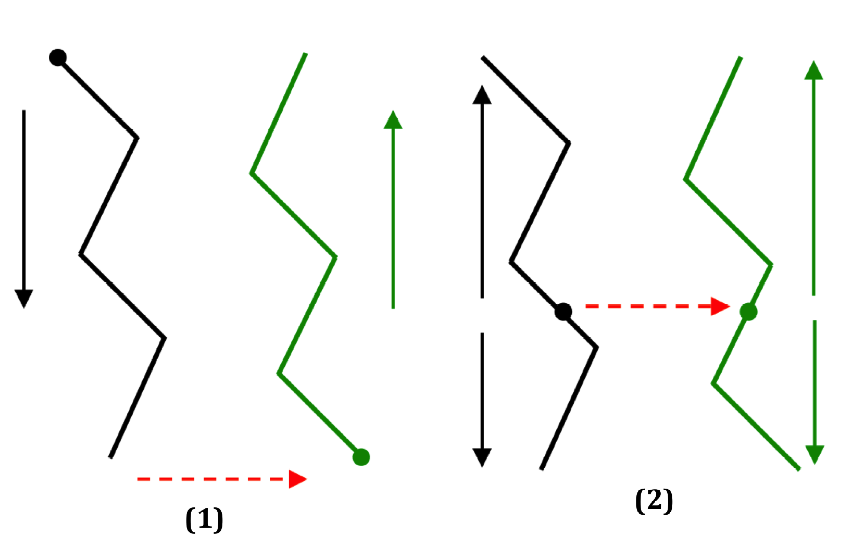
The dashed red lines show the jumps and the arrows show the direction of each edge.
Now, let's run again, but this time with the new fold tree we just defined:
$> ../../../main/source/bin/rosetta_scripts.default.linuxgccrelease -s inputs/ubq_dimer.pdb -parser:protocol inputs/ubq_relax2.xml -out:prefix test2_Take a look at the test2_ubq_dimer_0001.pdb output and compare it with the original one. You can see that the drastic movements of chain B due C-terminal relaxation of chain A is now diminished.
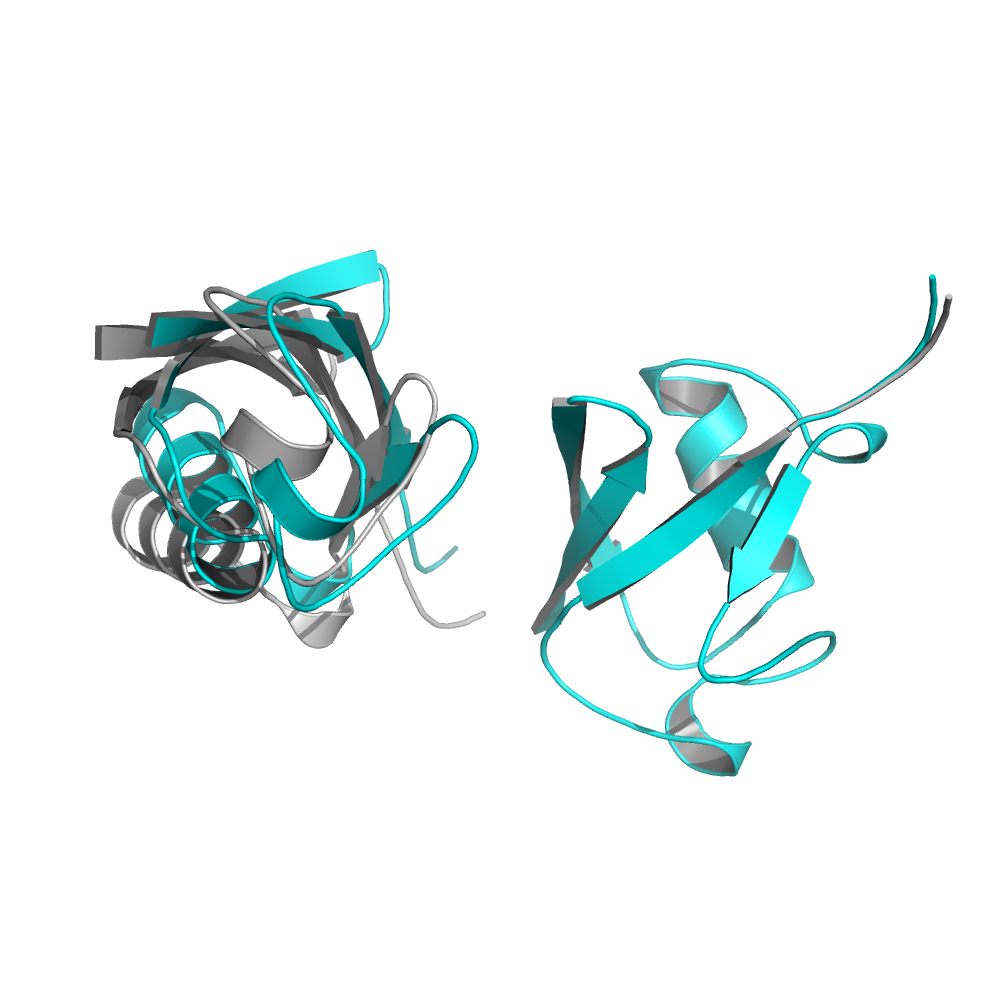
The original structure is in grey and the test2 structure is shown in cyan.
Final Points
Now you know what a fold tree is and how it is used to control the movements in your structure and how to control it. You can use fold tree to also mention how different chains in a structure are supposed to move with respect to each other. For example, if your chain C is between chain A and B in a complex and its movements should affect chain B, you can use a fold tree that places chain C downstream of chain B. The same principles apply for any chains, including a ligand or a metal. So, you can control the behavior of their movements by applying different fold trees. If you are interested in fold tree set up for a symmetric pose, please check here. Please note that a fold tree must contain NO CYCLES. In other words, you cannot define a residue both as upstream and downstream of other set of residues.