rosetta_basics/HBond Preferences Of Buried Polars
Note: Brian doesn't know where to put this data. At least here it will be public on the web.
Author: Brian Coventry 2019
Description
This great site: Atlas of Side-Chain and Main-Chain Hydrogen Bonding, collected statistics on the h-bonding preferences of buried polar atoms. This information is incredibly useful, but unfortunately, it was created in 1993.
With the PDB much larger than it was in 1993, a similar analysis was performed on Rosetta-relaxed version the PISCES dataset[1].
Criteria
In order be considered buried, the following must be true of an atom:
With a 0.9Å probe radius, the atom (and its attached Hs) must have 0 SASA
Using molecular surface atomic depth[2][3], the atom must be buried to at least 5.5Å
For the "entire group" graphs, every atom of the group must pass the above two criteria in order to be counted.
In order to be considered h-bonding:
- Rosetta beta_nov16 must classify an h-bond having at least -0.25 kcal/mol energy*
*Note: the bb_donor_acceptor_check() was set to False
HBond Preferences
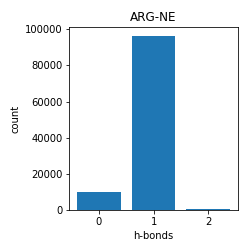
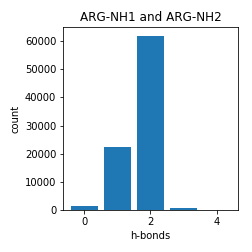
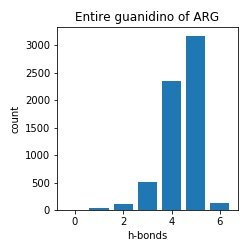
Arginine
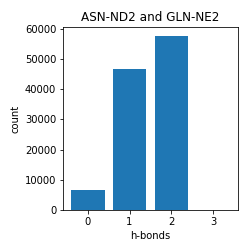
Asparagine and Glutamine
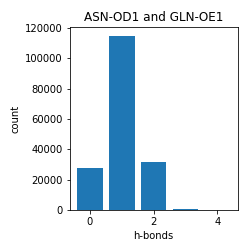
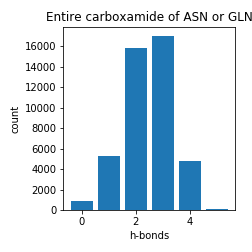
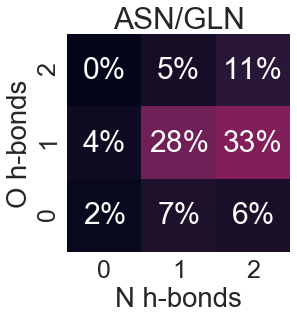
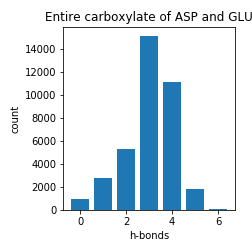
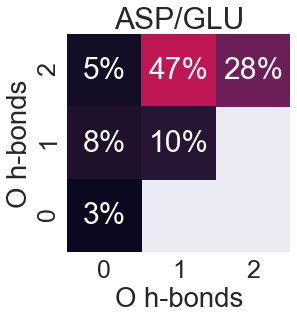
Aspartate and Glutamate
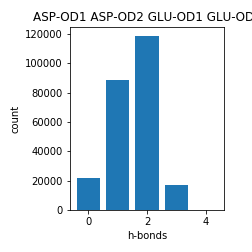
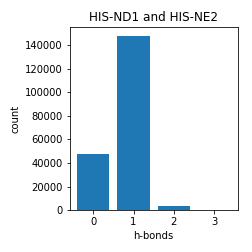
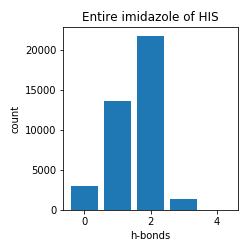
Histidine
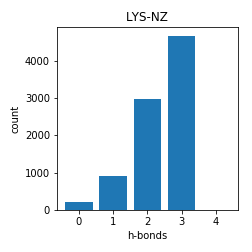
Lysine
The data for 0 h-bonds here is suspect. The code wasn't properly written to identify cross-linked lysines and often during the Rosetta-relax, rosetta decided to break lysine h-bonds.
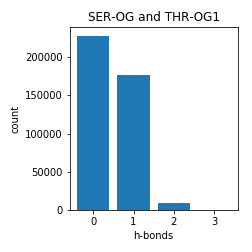
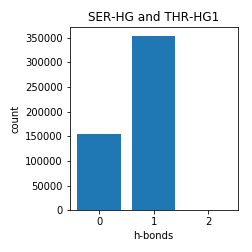
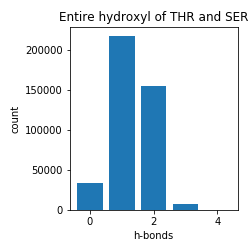
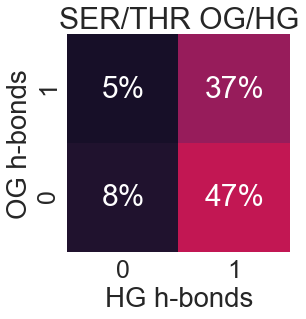
Serine and Threonine
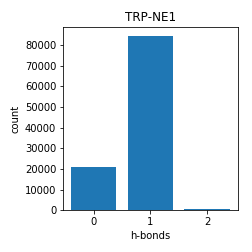
Tryptophan
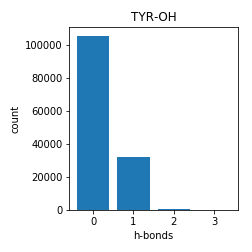
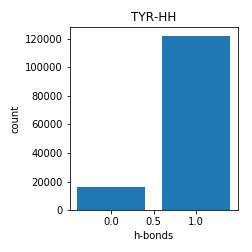
Tyrosine
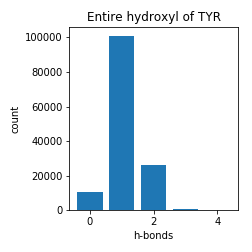
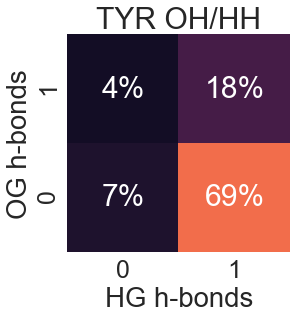
Notes
These numbers must be interpreted with care. While these show the number of h-bonds seen in nature, the maximum in these graphs does not necessarily coincide with the ideal number of h-bonds for "satisfaction". One must remember that adding another h-bond to a polar atom is a geometrically constrained problem; degrees of freedom are lost when setting up a protein to make another h-bond. For this reason, the number of h-bonds for "satisfaction" is likely higher than the numbers presented.
For the reason noted above, one cannot calculate the energy of a "buried unsatisfied hydrogen bond donor/acceptor" directly from this data. One must factor in the difficulty of making an h-bond to an atom. Which likely varies by side-chain type and number of h-bonds already present.
Citations
G. Wang and R. L. Dunbrack, Jr. PISCES: a protein sequence culling server. Bioinformatics, 19:1589-1591, 2003.
D. Xu, Y. Zhang (2009) Generating Triangulated Macromolecular Surfaces by Euclidean Distance Transform. PLoS ONE 4(12): e8140.
D. Xu, H. Li, Y. Zhang (2013) Protein Depth Calculation and the Use for Improving Accuracy of Protein Fold Recognition. Journal of Computational Biology 20(10):805-816.